After the post below regarding the
Alexa Dye conjugations, I had a few questions from users, not about the Alexa Dyes available, but about the titration, and choosing the optimal antibody concentration. Titrating your antibodies will go a long way towards achieving good quality flow cytometry data. A properly titered antibody will allow you to achieve the optimal separation between positive and negative without unnecessarily wasting antibody. This should appeal to you in multiple ways; better data, fewer experiments, and saving money. Below I will share with you the UCFlow, fool-proof method for antibody titration.
The first things to look at are, what's your antibody, which cells should I use to titrate, and in which state do the cells need to be for maximal staining? The 1st one's a no-brainer, but what you should be aware of is on which instrument (i.e. lasers and filters) you'll need to run this antibody:fluorochrome pair. Other things to be aware of is what is the species, specificity, and isotype of the antibody in question. This may impact what type of blocking may be necessary. For example, a rat anti mouse IgG2a may require the use of an anti-Fc receptor blocking antibody (for example 2.4G2) prior to staining in order to minimize non-specific binding of the native Fc receptors by the rat antibody. Another example would be if you needed to stain human blood cells with a rabbit anti-human Ig. In this case, you may pre-incubate the cells in normal rabbit serum to avoid non-specific staining. Next, we'll need some cells on which we'll titrate the antibody, and we'll need to know in which state these cells need to be in order to achieve max expression of the antigen. Take, for example anti-mouse CD44. CD44 is expressed on multiple cell types (bone marrow myeloid cells, peripheral T cells, etc...), and at different levels on the same cell type (upregulated on peripheral T cells), so you want to make sure there is enough antibody to sufficiently stain the highest expression level without using so much antibody that your background is too high. So, in a case like this, it may be better to either use mouse bone marrow, or perhaps activate peripheral lymphocytes to achieve the highest expression level you may encounter.
Now that we know what and with what we will test, we can proceed to the actual testing. If forced to take a stab in the dark at an optimal antibody concentration for flow cytometry, I'd guess 1ug of antibody in a final volume of 100ul of anywhere from a couple hundred thousand to a couple million cells. With this information, I generally start my titration 10 times that value, and stain my 1st sample with 10ug of antibody in 100ul of cells ( i typically use 500,000 cells, but that's not as important as keeping the volume of the sample consistent). From there, I do 3-fold serial dilutions down to 0.005ug, which gives a total of 8 tubes. I do all the staining in a 96 well plate, so each antibody being tested conveniently fits in 1 column. So the math pretty much goes like this. Take 15ul of the antibody (if stock is at 1mg/ml concentration) and put it into A1. Put 10ul of buffer into A2 - A8. Take 5ul from A1, mix into A2 (thereby creating a 3-fold dilution) and repeat serially down the plate. Be sure to remove the 5ul extra from A8 and discard. So, each well has 10ul of the properly diluted antibody. To each well, I add 90ul of previously blocked cells (remember, you were suppose to block the Fc receptor with 2.4G2 - this can be done in bulk prior to putting the cells into the wells). Incubate, 15 minutes at 4C, wash 2x with PBS + 1%BSA, and resuspend in 250ul of PBS +1%BSA. Also be sure to save a small amount of the cells alone (i.e. unstained) for setting up the instrument. Set up the cytometer with the appropriate lasers and filters, and collect data files. You may ask yourself, what if my stock antibody is at a lower concentration, or, what if I'm using an antibody with unknown concentration because the vendor only told me to use 1ul per "test." First, call the vendor and demand the concentration, they may not give it to you, but if enough people complain maybe they'll get the picture that real scientists like to work with known numbers not arbitrary ones like "tests." Secondly, if you are told to use 1ul per test, you can pretty much assume that's way too much antibody (since they want you to waste antibody so you'll buy more), so maybe my top concentration would be 3ul/test, and do a 3-fold dilution from there.
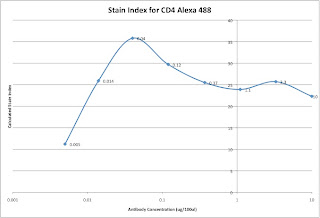
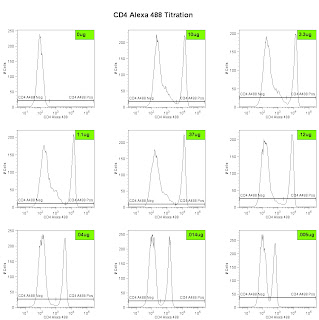
Lastly, we can analyze the data and generate the metric called the Stain Index. Perform standard gating of the population of interested (e.g. lymphocyte gate, live gate, myeloid gate, etc...). For the antibody titration channel, you will need to set a gate based on the 10ug sample which best separates the data into positive and negative groups. You'll then need to move the gates down as the positive population gets dimmer (see Titration Image). You can try to use the magnetic gate feature, but it doesn't always work that well. Once you have the positive and negative populations gated, you'll want to request the Standard Deviation of the negative population and the Median Fluorescence Intensity (MFI) of both neg and pos. The stain index can then be calculated as (MFIpos - MFIneg)/SDneg. A scatter plot of Log Antibody Concentration versus Stain Index should yield a curve whose max Stain Index is the optimal concentration (See Stain Index Image).
And there you go, a very easy way to get to the ideal concentration of antibody for flow cytometry. Remember, the concentration is critical, so we always try to stain in a final volume of 100ul. If you need to stain large numbers of cells, for example if you are sorting, then you'll need to increase the volume, and the concentration as well (probably never more than 1mL, though). If you plan to do this routinely, then a smaller scale titration (3 or 4 tubes instead of 8) may be necessary.




