I've been doing a good amount of application development recently and have had to "practice what I've preached." Those of us in the flow cytometry world, especially those in core facilities, like to pontificate all the do's and don'ts of flow cytometry, but how many of us have (recently) struggled through all the intricacies of perfecting a staining assay. I must say, I was a bit cavalier when I first agreed to set some protocols up for an investigator. The staining protocols weren't anything novel or difficult, it's just that I personally had not done some of the assays in quite a while. As I was going through the process I thought, hey, this is not as trivial as one might think...and I've been doing this for a loooooong time. I could only imagine what someone who is brand new to flow cytometry as a technique must feel like when their PI suggests they use this technology to investigate their hypothesis. So, I can put forth my top 10 steps to a successful flow experiment with some conviction, because I have now walked in your shoes.
I really wanted to make this a top-10, but as hard as I tried, I could only pare things down to 11. So, without further adieu I present to you;
10 11 Steps to a Successful Flow Cytometry Experiment
1.
Read lots of protocols (not just the reagent manufacturer's protocol). Let's face it. If you ask a dozen people how to make a peanut butter and jelly sandwich, you'll end up with 12 different recipes. The same goes for FCM protocols. Everyone finds a different part of the protocol worthy of emphasis. If you read a few of them, you can start to put the entire story together.
2.
Know which colors work best on your instrument. This is probably a bigger deal when you're using a core facility with a few different platforms. Let me tell you firsthand, no two cytometers are alike in their capabilities, not even two of the same model of cytometer. If you're lucky enough to have a flow cytometry core with knowledgable staff, make sure to ask them what their favorite 4, or 5, or 6-color panel is. They should also be able to tell you what the limitations of certain colors on a given instrument may be.
3.
When designing your panel, look for max brightness with min spillover. Ok, let's say you know what sort of antibodies you want to run, and you know what's available, as far as hardware goes, at your institution. Now comes the fun part. You have a list of antibodies, and a list of fluorochromes - how do you match them up? You've probably heard the old adage, put your dim fluorochromes on the antibody that targets abundant antigen, and your bright fluorochromes on antibodies against sparse antigen. In addition to that you want to minimize spillover - fluorescence from probes that are excited by the same laser and whose emission overlaps. Spillover = Background, and Background = Diminished resolution. This takes some effort and a bit of know-how, so consult your friendly flow guru for help, or try out some of the new utilities designed to help with this process (namely
CytoGenie from Woodside Logic or
Fluorish from Treestar).
4.
Titrate your reagents. What for? The manufacturer told me to use 5ul per test (usually 10^6 cells in 100ul of volume). Without jumping on the conspiracy theory bandwagon that reagent manufacturers tell you to use too much antibody so that you'll waste your antibody and have to buy more, I will say that I've found more times than not that the manufacturers suggested dilution is too concentrated. If you want to see why you should titrate your antibodies, check out the figure below. If you want to see how to titrate your antibodies, click on over to this
prior entry to the UCFlow Blog.
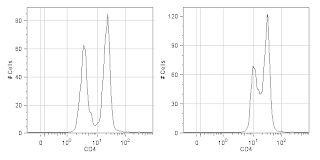 |
CD4 staining of fixed human PBMCs at the properly
titrated concentration (Left) and the manufacturer's
recommended concentration (Right).
|
 |
| Example Staining Worklist |
5.
Outline your plan of attack. Make a detailed work list of your protocol. Generic protocols are good to help plan your experiment, but when it comes time to perform the steps of an assay, you really want a work list. As the name implies, this is a step-by-step recipe of how to execute the protocol. I usually include the step, duration, volume of reagent, temperature, etc... While you're performing your assay, take copious notes so you can fine-tune the protocol, adding more detail. The goal is to be able to hand this work list and the reagents to another user and they should have successful results. I like to do this in Excel and write in all the cell formulas so that I can type in how many samples I need to stain and have it automagically do all my dilutions for me. I also have a summary of the buffers needed and quantities at the bottom. See below as an example.
6.
Always use a Dead Cell Marker. Dead cells can really screw up an analysis. I guarantee there is a color and assay compatible dead cell marker available for most every experiment you will do. There's no excuse not to use a dead cell marker, so please, please do it. It makes for a much nicer looking plot, and you really can't do good (dim) double positive enumeration without it.
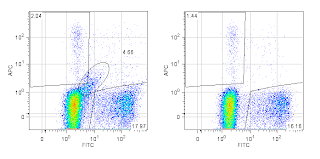 |
Two-parameter plot without using an upstream dead cell
marker (Left) and the same plot after removing dead
cells (Right). Note the diagonal population extending
out of the negative population (encircled with a region
in the left plot)
|
7.
Set up your FMO's as a separate experiment, not on your real samples. I won't discuss the merits of using an FMO control (
Fluorescence
Minus
One), let's just assume you know that it's pretty much a necessity. What I will say is if you try and set up an FMO control on the day that you're using your precious sample, you're likely to either forget it, or omit it because you think you don't have enough cells. So, if possible, set up your FMO controls ahead of time on a different day so you can take your time getting everything set up properly. It'd be nice to include it every time, if you have enough sample.
8. M
ake compensation controls using beads. I'm a huge advocate of using capture beads to set up compensation. It's really a no brainer. I've written about
this subject before. Even if your single stained controls look fine on cells, I'd still use beads because they're always consistent.
9.
Acquire your samples nice and slow to achieve maximum resolution. If you go through the trouble of perfecting your staining procedure, now's not the time to screw things up. On a hydrodynamically focused instrument you'll want to concentrate your sample and run it slow in order to keep a narrow core stream and achieve optimal resolution. If you're using another type of flow cell (such as a capillary a la Millipore or an acoustically focused system like the Attune) you should be more focused on increases in background due to insufficient washing rather than a wide sample core.
10.
Analyze your data a couple of different ways. Even if I have a clear idea of how to go about the analysis, I'm frequently surprised at how many times I've changed axes or started backwards and found I liked the new way better than the old way. Backgating is one way to help identify a rare population all the way up through its ancestry. Make sure to take advantage of your Live cell channel as well as gating out aggregates and removing any time slices where there may have been a drift in fluorescence.
11.
QC your instrument and create an application specific QA protocol. Science is not about 1-shot deals. If it's not reproducible, it's not real. In order to give you the best possible chance of getting reproducible data you'll want to minimize the error contributed by the instrument. Quality control and Quality assurance cannot be emphasized enough. By doing something as simple as running beads at your application-specific voltage settings you can ensure that the instrument is in the same state as it was the last time you acquired these samples. For this, I typically use one of the peaks (peak 4, actually) of the 8-peak bead set. After I have the samples acquired with the proper voltage settings, I run the beads, create target channels for the peaks and save it as a template. Next time, all I need to do is dial in the voltage to put the beads in the target. You'll also want to make an Acquisition template and probably an analysis template too.
Well, there you have it. Hopefully this will help you focus your attention on some key aspects of setting up a well-thought-out flow cytometry staining protocol. Of course, this merely scratches the surface of all the things you need to think about. Did I miss something major? Feel free to leave a comment with your #12, #13, and beyond.




