I've been composing this post in my head for a couple of weeks now, but have been too busy to sit down and write it, so it shouldn't really surprise me that it popped up in a dream. However, a new twist was added via my unconscious mind (which I'll get to later). So, the original post was all about how I've pretty much given up on compensating using cells, and if you're not using beads, then you're pretty much setting yourself up for compensation failure (unless of course you're using things like PI, or mCherry, or the like). I mean, the whole point of 'autocomp' is to take the subjectivity out of compensation, and using objective mathematics to correct for fluorescence spillover. However, every single time I've done autocomp using cells, it just doesn't look 'right' and I end up tweaking the values just a little bit. I've come to terms with this fact, and have pretty much settled with this sub-par situation. But, if you're trying to teach someone about compensation, and you introduce this 'autocomp' feature, it makes for a pretty awkward conversation when you then go on to say, "Well, just adjust the values a little bit until it looks right." So, I typically recommend people do their compensation with beads. For many of my users, the thing that prevents them from doing this is cost, or maybe a bit of skepticism in changing the ways they were taught to do their staining. The reasons why compensating using cells doesn't always work are many, but let me just outline a few for you here.
1. Insufficient frequencies of both positive and negative fraction to make a statistically significant regression of means. If in your stained cell sample, you only have a 0.1% positive fraction, the mean of that population in the spillover channel will not reach a high enough statistical significance until you collect millions of cells. No one is going to collect millions of cells on their single stain control. This also holds true when all your cells are positive for your single stain control, and you have a really low negative (or low) population.
2. Poor resolution of the positive fraction. Sometimes you will not have a clear positive population, so making a gate around the positive fraction for performing compensation is difficult. If you end up encircling some of the high autofluorescent cells that you mistakenly call positive, your compensation will surely be off.
3. Non-linearities at the extremes can lead to inaccurate compensation. If you're compensating using an unstained (or negative) fraction that is at the very low-end of the scale, or if your positive fraction is at the very high-end of the scale, you're likely using a data point in the non-linear range of the log scale. Since compensation algorithms are basically relying on the fact that your range of analysis is linear, you're going to run into lots of problems if you're using "unstained" cells as your low-data point, or really bright cells as your high data point. Side bar: Yes, I know, your comp control should be at least as bright as your sample staining, blah, blah, blah. However, the only reason why this is the case is because of non-linearities at the very high end of the scale. If all your staining fell within the linear portion of the scale (let's say 1.0 logs to 3.5 logs), then this isn't necessarily a problem. You can take any two points within that range, and create a regression line that will model the entire scale. No-scale is linear enough, especially at the extremes, so the 'rule' of a maximally bright comp control needs to be adhered to.
4. Mismatched autofluorescence between positive and negative. If I stained my leukocyte prep with a monocyte marker (CD14, for example). All my monocytes will be positive. For this single stained comp control, what should I use as my negative? Many people would simply use the negative lymphocytes or granulocytes, and many people would end up with a poor compensation matrix. For channels where autofluorescence is a factor (mostly the green/yellow detectors off the blue and lower laser lines), the positive fraction's autofluorescence should match the negative fraction's autofluorescence. This is, evidently only necessary when you're using cells for compensation, and you have a mixed cell-type sample.
So, there are certainly lots of pitfalls when using cells for compensation, which is why using beads is a good idea. To solve many of these issues, simply using an antibody capture bead at two fluorescence levels should do the trick. You'll notice I said two fluorescence levels, and not one positive and the 'blank' bead. Using the blank bead can lead us into issue #3 above, so I prefer to use the bead at a saturating level of antibody and maybe 100-fold less, to create a high and low peak. In the end the peaks will fall around the 3.5 decade range and 1.5 decade range. Use these peaks as your 'positive' and 'negative' values in your favorite autocomp program, and voila, perfect compensation. Of course, these beads are run at the appropriate voltage that is set up according to your cell type.
But, what about the twist? The twist is, that you don't need to only use beads as your capture matrix. You could use cells! I know, I know, I just went on and on about NOT using cells, now I'm telling you to use cells, but wait, let me explain. Take a thymus, get all your non-tandem antibodies in CD4, stain them at two concentrations, fix them, and stick them in the fridge. You now have ready-made compensation controls that are much cheaper than buying capture beads. Why thymus? They're the closest thing to beads; pretty much homogeneous, so we don't have to worry about autofluorescence mismatch, they're almost all CD4 positive, so that makes it easier to create two nice peaks, and you can get a boatload of them from a young mouse. On top of all this, we gain the ability to use other things besides antibodies. You could stain them with many of your dyes for a comp control, PI, DAPI, CFSE, etc... Something you can't do with beads. For tandems, I'd stick with capture beads.
Ok, there you have it. If you've made it this far reading through all my gibberish, let me know what you think.
Tuesday, November 9, 2010
Friday, October 1, 2010
MoFlo Upgraded to XDP, plus a couple new laser lines.
Ah, the MoFlo - what a fine piece of craftsmanship! I started my relationship with the MoFlo (Formerly of Cytomation, Formerly of DakoCytomation, Formerly of Dako, Currently of Beckman-Coulter) in the year 2000. We had many great years together, but our relationship was getting a bit stale. You see, there was this fancy new gal in town call the Aria who lured me into her web of seduction with promises of 'turn-key' operation, and I bit! I soon realized however, that the grass isn't necessarily greener on the other side, and re-visited the rock-solid usability of the MoFlo. In recent years, the MoFlo started showing its age. I have to admit, part of the issue was a certain level of neglect and abuse on our part, but hey 10 years in instrument years is like 80 in people years. And so we came to a fork in the road, and as with most things in the technology area utilizing 20 year old components, we had to decide, pull the plug or pursue the upgrade path.
When I was contacted by the folks at Propel labs (who, evidently are a group of people from the original Cytomation company) that there was an upgrade path to the XDP electronics for the legacy MoFlo, I was thrilled. After about a year of begging for money from anyone that would listen to me, I finally secured the funding and was ready for the upgrade. So, why upgrade to XDP instead of buying a new sorter? Well, first of all, it was a financial thing. The cost of an upgrade is about 1/4th the cost of a new sorter. Secondly, the fluidics on our MoFlo are uncannily stable; who knows if we'd strike it rich again with a new sorter. You may also be asking, what's so great about XDP? Well, I'd never be able to explain with such elegance as Dan Fox could, so all I can say is track down the white paper Dan wrote, read it, then pick your lower jaw up off the floor. The big lure for me (besides the obsolescence of parts for the legacy MoFlo) was just the fact that we'd be able to operate with no/low hard aborts similar to the Aria, which, when paired with the higher number of droplets a jet-in-air can achieve, should allow us to sort faster and maintain high yields and purity. With our XDP upgrade, we also had all our PMTs changed, and threw on two new laser lines to boot. - Side note - We had one of these co-lase towers installed on our MoFlo, which is also a product of Propel Labs, that basically combines two laser lines so they can be run colinear into the 3-pinhold MoFlo setup. We chose to put on a UV and Red laser and run them colinear through the co-lase tower. This now gives us a 4-laser MoFlo (355, 488, 561, and 640) - End Side Note -
As far as the actual upgrade goes, the install went pretty smooth. It took 2-3 guys about 3 days to completely tear down the instrument to basically an empty table, and then install the PMTs, electronics, the touch-screen panel, and the computer. As with most installs/upgrades, we did have a couple hiccups, but they were taken care of immediately. I guess that's one good thing about working with a smaller company like Propel Labs. They can't afford to lose any business, so customer service is automatically very good.
We've been using the XDP now for about a week, and things have gone pretty well. We're still getting use to the touch-screen interface, and some of the new things in Summit, but overall, I'd say we made the right decision, and hopefully the MoFlo can dutifully give us another 10 years of service.
Once we've gotten into a rhythm on this thing and really test the bounds of speed, I'll post some data. But for now, enjoy a pic of the finished product below.
When I was contacted by the folks at Propel labs (who, evidently are a group of people from the original Cytomation company) that there was an upgrade path to the XDP electronics for the legacy MoFlo, I was thrilled. After about a year of begging for money from anyone that would listen to me, I finally secured the funding and was ready for the upgrade. So, why upgrade to XDP instead of buying a new sorter? Well, first of all, it was a financial thing. The cost of an upgrade is about 1/4th the cost of a new sorter. Secondly, the fluidics on our MoFlo are uncannily stable; who knows if we'd strike it rich again with a new sorter. You may also be asking, what's so great about XDP? Well, I'd never be able to explain with such elegance as Dan Fox could, so all I can say is track down the white paper Dan wrote, read it, then pick your lower jaw up off the floor. The big lure for me (besides the obsolescence of parts for the legacy MoFlo) was just the fact that we'd be able to operate with no/low hard aborts similar to the Aria, which, when paired with the higher number of droplets a jet-in-air can achieve, should allow us to sort faster and maintain high yields and purity. With our XDP upgrade, we also had all our PMTs changed, and threw on two new laser lines to boot. - Side note - We had one of these co-lase towers installed on our MoFlo, which is also a product of Propel Labs, that basically combines two laser lines so they can be run colinear into the 3-pinhold MoFlo setup. We chose to put on a UV and Red laser and run them colinear through the co-lase tower. This now gives us a 4-laser MoFlo (355, 488, 561, and 640) - End Side Note -
 |
| The remains of the MoFlo after the tear-down |
We've been using the XDP now for about a week, and things have gone pretty well. We're still getting use to the touch-screen interface, and some of the new things in Summit, but overall, I'd say we made the right decision, and hopefully the MoFlo can dutifully give us another 10 years of service.
Once we've gotten into a rhythm on this thing and really test the bounds of speed, I'll post some data. But for now, enjoy a pic of the finished product below.
 |
| The upgraded MoFlo XDP in all its polished glory! |
Thursday, September 23, 2010
Follow us on Facebook!!

Find the University of Chicago Flow Cytometry Core Facility group and feel free to leave any comments and questions you might have.
Wednesday, September 22, 2010
GLIIFCA Core Manager Meeting Preview - September 24, 2010
For those who will be at the meeting, I've put up my slides in PDF format here (sorry, I think the link works now) in case you wanted to follow-up with one of the tools we use in the core. For those who will not be able to attend, feel free to read through to see what types of tools we use at UCFlow to try to do more with less and be as efficient as possible. Check back here for updates from the Core Manager meeting and the rest of this year's GLIIFCA.
Thursday, September 16, 2010
Flow cytometry leads to everything!

The man on the right in this picture is Jeff Schneider. He used to be a technician here in the flow lab. Look at him now, playing with his band Darling at the Hideout tonight at 9PM. Rock on dude!
Flow cytometry leads to everything.
Friday, August 27, 2010
You know what really grinds my gears?
So, I was fixing a clogged DCM pump on the LSRII this morning, which requires the removal of the side panel on the instrument, getting on your hands and knees and digging around in the inner bowels of the beast. As I was shimmying around, contorting my limbs in all sorts of god-awful positions, I kept crunching pieces of plastic under my feet. I peaked down, backwards, over my shoulder to see a bunch of pipette tips on the floor...surrounding the trash bin at the foot of the instrument. Are you kidding me? People, presumedly adding their PI or DAPI at the instrument, are ejecting their used tips in the vicinity of the garbage, and missing >50% of the time. They obviously can hear the tips crashing into the ground and missing the garbage, but decide to do nothing about this? I then pan across the room, and I see racks of nearly empty tubes, crumpled kimwipes and a full waste tank. C'mon people, have a little respect, pick up after yourselves. Needless to say, my DCM repair turned into a full-on cleaning session; swept, threw out all the tubes, wiped down the bench space, and even got the mop from the janitor's closet and mopped the floors. So, here we go people, August 27th, 2010 at 10AM, the LSRII area in 037 is clean. LET'S KEEP IT THAT WAY!
Thursday, August 12, 2010
It's GLIIFCA Time!
We're in the dog days of Summer, so that could only mean one thing... GLIIFCA 19 is right around the corner. The Great Lakes International (because we let the Canadians join us) Imaging and Flow Cytometry Association's 19th annual meeting will be held September 24 - 26, 2010 in Detroit, MI. This is a great meeting for users of Flow Cytometry and Imaging technologies (including the ImageStream!) that gives you a chance to see how people are using Flow in interesting and novel ways in their research. It's also ridiculously cheap. The registration fee is a paltry $80 AND, if you bring a poster, you'll get a $100 travel stipend. Also, it's close enough to drive, and the hotel rates are very reasonable as well. So, there's really no excuse not to go. However, if you need one more reason why you should go to GLIIFCA this year, it's because the theme for this year's Saturday night party is freaking STAR WARS!!!! So, click on over to GLIIFCA.org and register yourself today. If you U of C folks do decide to join us, David will buy you a beer!
Friday, July 16, 2010
Got Sand?
Or should I say, do you need sand, because I have some, well, a bunch of sand in my office! It has sort of turned into a UCFlow tradition now, but the office pranksters have done it again. I'm not sure why I was surprised since this happens pretty much every July to me, but I have to admit... they got me good this time. If you have no clue what I'm talking about let me explain. Every time I leave the lab for an extended period of time (vacation, meetings, etc...) I come back to find my office 'decorated' with the latest musings of lead prankster, David Leclerc. Now, these aren't just, "let's put a whoopee cushion on his chair" types of pranks, these take coordination and time, lots of time (I'm sure it all takes place after 5PM ; ).
In prior years, I've come back to find my office wallpapered with post-its, which sounds sort of crazy, but add to that, the fact that every single post-it had a little nonsensical note written on it. Next was balloons. My office was filled to about waist height with balloons - luckily they were filled with air, and not water or other types of noxiousness. The next year was aluminum foil. My office and everything in it was wrapped in foil. This was pretty extreme in that even the few pennies I had sitting on my shelf were individually wrapped in foil. Which brings me to this year's vandalism. As I approached my office on Monday morning, I started to notice a bit of graininess on the floor, still puzzled, I opened my door to find a "tropical paradise." There was about 3 inches of sand covering the entire floor, a "palm" tree, empty bottles of corona, sand toys, and even a sand sculpture of the Eiffel Tower.
So, I haven't figured out exactly how I'm going to clean this up. Luckily they were smart enough to put a plastic tarp over my carpet, so that should help. But, seriously, if you are putting down some patio pavers this summer and need some sand, let me know!


In prior years, I've come back to find my office wallpapered with post-its, which sounds sort of crazy, but add to that, the fact that every single post-it had a little nonsensical note written on it. Next was balloons. My office was filled to about waist height with balloons - luckily they were filled with air, and not water or other types of noxiousness. The next year was aluminum foil. My office and everything in it was wrapped in foil. This was pretty extreme in that even the few pennies I had sitting on my shelf were individually wrapped in foil. Which brings me to this year's vandalism. As I approached my office on Monday morning, I started to notice a bit of graininess on the floor, still puzzled, I opened my door to find a "tropical paradise." There was about 3 inches of sand covering the entire floor, a "palm" tree, empty bottles of corona, sand toys, and even a sand sculpture of the Eiffel Tower.
So, I haven't figured out exactly how I'm going to clean this up. Luckily they were smart enough to put a plastic tarp over my carpet, so that should help. But, seriously, if you are putting down some patio pavers this summer and need some sand, let me know!


Wednesday, June 23, 2010
You mean there's other analysis software besides FlowJo???
Even though a vast majority of users at U of C are FlowJo users, it doesn't mean we ONLY use FlowJo. FlowJo does a really good job with a lot of different flow applications, but there are some things I'd definitely want to change. So, I'm always playing around with new software, as well as revisiting old standbys: FCS Express, VenturiOne, Kaluza, Winlist, to name a few. In addition, there are a bunch of freeware apps that are pretty good as well: WinMDI, Cyflogic, MFI, ANALYSE, IDLYK, and more being developed frequently. Recently I had the opportunity to talk with Kelly Rae Chi from The Scientist Magazine, which resulted in an article featured in the most recent edition. The piece is titled "Let the Data Flow: Rethink your Data Analysis Tools for Flow Cytometry." It's in Volume 24, Issue 6, Page 63.
Wednesday, June 9, 2010


Thursday, June 3, 2010
Intellicyt HyperCyt Impressions
Well, the HyperCyt has come and gone and to sum up my thoughts in a word I can only say, Wow. When it comes to speed, this thing is really fast. I tried to put a bunch of different things through the sampler to try and get it to fail, but it just didn't; big stuff, clumpy stuff, small stuff, dyes, you name it.
Now, let me put things into perspective for you a bit. We run everything in tubes manually, so to run through 96 samples I've been able to achieve a throughput rate of about 40 minutes (collecting a couple of thousand cells per sample). My target for a sampler was to get an equivalent amount and quality of data in less than 20 minutes per plate. By many accounts, this is NOT high throughput. The representative from Intellicyt actually sort of chuckled at me when i was explaining this. His idea of high throughput was collecting 384 well plates in about 6 or 7 minutes. I certainly can appreciate the utility of this unit for something like that, but my standards of data quality are probably a bit more strict when compared to a "screening" assay.
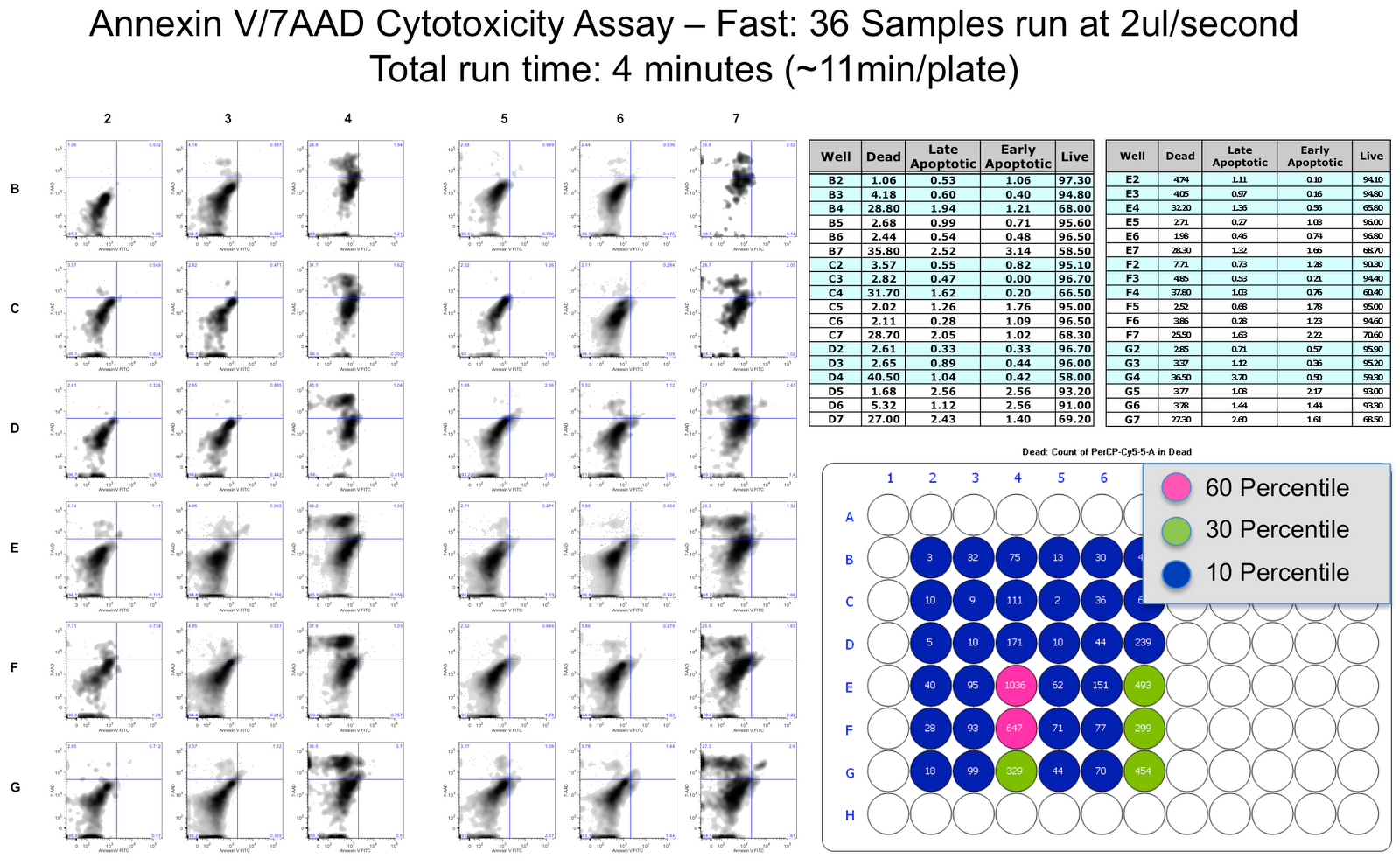
What I was really impressed with was the ability to precisely control every aspect of the sampling process such that you can go really fast, or pretty slow if you wanted to. What I am showing here is a comparison of the same exact plate run at a "high" speed (~11 minutes per 96 well plate) and at a "low" speed (~27 minutes per 96 well plate). It is a simple Annexin V FITC vs. 7-AAD cytotoxicity assay, that is comparing the affects of a drug on the cells. As you can see, there is no appreciable difference in the two, and they both are the same as when I ran these samples manually in a tube.
 Other things I ran were two big cell lines (one GFP, one RFP) mixed together to look at the potential increase in coincidence events (double positives) when going at high rates. When I ran the sample manually in a tube, I saw about 0.5% double positive cells. When I ran them through the HyperCyt at the higher flow rate, that double positive population shot up to 1.6%, and when I ran it at the slower flow rate, it was about 1%. The reason this happens, as I'm told, is that when the unit sends the bolus of cells into the cytometer, the cells tend to accumulate at the leading edge of the bolus, so you get a high concentration of cells right in the beginning of collection, thus leading to higher coincidence. In fact when I looked at the formation of coincident events versus time of a single collected sample I saw exactly that, all the double positives were detected in the 1st second or so of collection, then after that, it leveled out to the 0.5% seen while running the samples manually in a tube.
Other things I ran were two big cell lines (one GFP, one RFP) mixed together to look at the potential increase in coincidence events (double positives) when going at high rates. When I ran the sample manually in a tube, I saw about 0.5% double positive cells. When I ran them through the HyperCyt at the higher flow rate, that double positive population shot up to 1.6%, and when I ran it at the slower flow rate, it was about 1%. The reason this happens, as I'm told, is that when the unit sends the bolus of cells into the cytometer, the cells tend to accumulate at the leading edge of the bolus, so you get a high concentration of cells right in the beginning of collection, thus leading to higher coincidence. In fact when I looked at the formation of coincident events versus time of a single collected sample I saw exactly that, all the double positives were detected in the 1st second or so of collection, then after that, it leveled out to the 0.5% seen while running the samples manually in a tube.
One other thing I was concerned with was the residual dyes left over when rinsing between samples. To test this, I ran alternating samples of PI stained CENs and DAPI stained CENs. I first ran it with no wash to show that the probe itself may be transferring dye from well to well (which it did). Then I ran the samples with a wash in between collection and then there was no increase in PI staining of the DAPI CENs and no increase in DAPI staining of the PI CENs. This, along with multiple other tests run with beads put me at ease as to the capabilities of this instrument as a multi-well plate sampler, not just a high throughput screen tool.
So, will I get one? Likely, but possibly in the form of a Accuri/HyperCyt combo ala what was displayed at the CYTO 2010 meeting. This is a new product from Intellicyt, which they're calling the HTFC (High Throughput Flow Cytometer). Here's a link to the info on that (warning, this is a link to a pdf, so it will automagically start downloading to your computer; not to worry though, it's safe).
Now, let me put things into perspective for you a bit. We run everything in tubes manually, so to run through 96 samples I've been able to achieve a throughput rate of about 40 minutes (collecting a couple of thousand cells per sample). My target for a sampler was to get an equivalent amount and quality of data in less than 20 minutes per plate. By many accounts, this is NOT high throughput. The representative from Intellicyt actually sort of chuckled at me when i was explaining this. His idea of high throughput was collecting 384 well plates in about 6 or 7 minutes. I certainly can appreciate the utility of this unit for something like that, but my standards of data quality are probably a bit more strict when compared to a "screening" assay.
What I was really impressed with was the ability to precisely control every aspect of the sampling process such that you can go really fast, or pretty slow if you wanted to. What I am showing here is a comparison of the same exact plate run at a "high" speed (~11 minutes per 96 well plate) and at a "low" speed (~27 minutes per 96 well plate). It is a simple Annexin V FITC vs. 7-AAD cytotoxicity assay, that is comparing the affects of a drug on the cells. As you can see, there is no appreciable difference in the two, and they both are the same as when I ran these samples manually in a tube.
 Other things I ran were two big cell lines (one GFP, one RFP) mixed together to look at the potential increase in coincidence events (double positives) when going at high rates. When I ran the sample manually in a tube, I saw about 0.5% double positive cells. When I ran them through the HyperCyt at the higher flow rate, that double positive population shot up to 1.6%, and when I ran it at the slower flow rate, it was about 1%. The reason this happens, as I'm told, is that when the unit sends the bolus of cells into the cytometer, the cells tend to accumulate at the leading edge of the bolus, so you get a high concentration of cells right in the beginning of collection, thus leading to higher coincidence. In fact when I looked at the formation of coincident events versus time of a single collected sample I saw exactly that, all the double positives were detected in the 1st second or so of collection, then after that, it leveled out to the 0.5% seen while running the samples manually in a tube.
Other things I ran were two big cell lines (one GFP, one RFP) mixed together to look at the potential increase in coincidence events (double positives) when going at high rates. When I ran the sample manually in a tube, I saw about 0.5% double positive cells. When I ran them through the HyperCyt at the higher flow rate, that double positive population shot up to 1.6%, and when I ran it at the slower flow rate, it was about 1%. The reason this happens, as I'm told, is that when the unit sends the bolus of cells into the cytometer, the cells tend to accumulate at the leading edge of the bolus, so you get a high concentration of cells right in the beginning of collection, thus leading to higher coincidence. In fact when I looked at the formation of coincident events versus time of a single collected sample I saw exactly that, all the double positives were detected in the 1st second or so of collection, then after that, it leveled out to the 0.5% seen while running the samples manually in a tube. One other thing I was concerned with was the residual dyes left over when rinsing between samples. To test this, I ran alternating samples of PI stained CENs and DAPI stained CENs. I first ran it with no wash to show that the probe itself may be transferring dye from well to well (which it did). Then I ran the samples with a wash in between collection and then there was no increase in PI staining of the DAPI CENs and no increase in DAPI staining of the PI CENs. This, along with multiple other tests run with beads put me at ease as to the capabilities of this instrument as a multi-well plate sampler, not just a high throughput screen tool.
So, will I get one? Likely, but possibly in the form of a Accuri/HyperCyt combo ala what was displayed at the CYTO 2010 meeting. This is a new product from Intellicyt, which they're calling the HTFC (High Throughput Flow Cytometer). Here's a link to the info on that (warning, this is a link to a pdf, so it will automagically start downloading to your computer; not to worry though, it's safe).
Core Fair Today!!!!
The Office of Shared Research Facilities is hosting it annual Core Fair today from Noon to 2PM in the GCIS atrium. This is your chance to visit with all of the ~25 different core facilities on campus to find out what's new and exciting in their labs. The flow lab will be present and is eager to share with you our new technology and services. We'll be highlighting the ImageStreamX, of course and all the neat things you can do with that instrument. But, we're also trying to spread the word about our Drop-Off Service and our forthcoming analyzer, the BD LSRFortessa. Last, but by no means least, we'll be letting people know about our exciting news regarding the MoFlo. (Drum roll, please) It has been a long time coming, but we've finally secured some funding to get our MoFlo upgraded to the MoFlo XDP package from Propel Labs. This will definitely breathe new life into our dying sorter.
Saturday, May 22, 2010
Qdots: they're not just for lighting up your antibodies any more
Get this; a company (spun-off from Fujitsu) has successfully made a green emitting laser using Quantum Dots. The company, called QD Laser, Inc. (how original) created a quantum dot semiconductor crystal that emits at 1064nm. A quick frequency doubling yields the 532nm laser line we all know and love. Now, they certainly aren't making this for the biomedical research crowd, but hey none of the tech we use today was developed for flow cytometry. It's the trickle down effect that gives us all the great gems. So, you're probably saying, who cares, a 532nm laser, we have a ton of them. Well, besides this being made from a Qdot, it also is super low-power consuming, and it is tiny; see the pic below. QD Laser envisions these lasers along with already available red and blue lasers as the projection source of an RGB miniature projector that can be embedded in smartphones. However, I'm envisioning these lasers on my handheld cytometer.


Wednesday, May 19, 2010
HyperCyt Demo 5/25, 5/26
We will be demonstrating a new 96/384 well sampler on the LSRII-Orange next week. To introduce the platform, we will host a seminar on Tuesday, 5/25 at 10AM in Cummings Room 119. On Wednesday the 26th, we are looking for people who wish to try out the sampler using their own samples. Since this unit will be installed on our LSRII-Orange, please be aware of the lasers/filters on that specific instrument. Here's a few stats on this system. More info can be found on their web site @: http://intellicyt.com/products_hypercyt.php
-Max speed: Full 96 well plate in 2.5 minutes, Full 384 well plate in 10 minutes
-Sample Volume: as little as 2ul with zero dead volume, and as much as a full well
-Can be connected to a variety of cytometers
-Carryover in High Throughput Mode can be 1-2%, but inter-well washes decreases carryover to less than 1%
If anyone else out there has one of these, I'd be interested in your thoughts as well. We've used the BD HTS system on a FACSCalibur with not-so-great results, so are looking for something more high throughput, and more stable.
-Max speed: Full 96 well plate in 2.5 minutes, Full 384 well plate in 10 minutes
-Sample Volume: as little as 2ul with zero dead volume, and as much as a full well
-Can be connected to a variety of cytometers
-Carryover in High Throughput Mode can be 1-2%, but inter-well washes decreases carryover to less than 1%
If anyone else out there has one of these, I'd be interested in your thoughts as well. We've used the BD HTS system on a FACSCalibur with not-so-great results, so are looking for something more high throughput, and more stable.
Tuesday, May 11, 2010
CYTO 2010
Here in Seattle at the CYTO 2010 meeting (the new branding of ISAC, for those unaware). It has been a very typical ISAC, content-wise, but a huge success, location-wise. Seattle has turned out to be a pretty ideal site for one of these meetings. We're having the meeting at the Washington State Trade and Convention Center in downtown Seattle. Being from Chicago, it sort of feels like home, but really, really condensed into about a 10-block radius. Literally, everything you need is in walking distance. So, Seattle definitely gets a thumbs-up from me.
As far as the meeting content, the 'hot' technology is definitely the CyTOF from DVS Sciences. We first saw this instrument at last year's GLIIFCA meeting, but there are now many good examples of the technology in the field...and it looks really nice. As you can probably gather from the name, it's a mass spec instrument that is being used to analyze cells. How? Pretty simply: you basically take traditional antibodies, and instead of coupling them to fluors, like FITC and PE, you couple them to mass spec-type stable isotopes. Then you load your 'stained' sample into the CyTOF, and ituses a laser to basically vaporizes and ionizes the compound in an Inductively Coupled Plasma (ICP-Mass Spec, not Maldi-Tof Mass Spec) yielding a specific signature. Since there are around 100 isotopes available for these types of applications, AND, the resolution between isotope detection, you can basically do a 50 'color' experiment without compensation. So, now, you can do huge multiplexed assays with really, really, good resolution. There are even examples of doing a luminex type assay on this. If you load the beads with a few different combinations of different isotopes, you can easily create a 1,000,000 plex luminex assay! I see an S-10 application in my future!
I'll update with more info again soon, including info about our posters!
As far as the meeting content, the 'hot' technology is definitely the CyTOF from DVS Sciences. We first saw this instrument at last year's GLIIFCA meeting, but there are now many good examples of the technology in the field...and it looks really nice. As you can probably gather from the name, it's a mass spec instrument that is being used to analyze cells. How? Pretty simply: you basically take traditional antibodies, and instead of coupling them to fluors, like FITC and PE, you couple them to mass spec-type stable isotopes. Then you load your 'stained' sample into the CyTOF, and it
I'll update with more info again soon, including info about our posters!
Tuesday, April 20, 2010
iCyt Acquired by Sony: FCM controlled by PSP Motion?
TVs, PSPs, PMPs, Laptops, and now Flow Cytometers? This seems like something out of left field, but Sony recently acquired University of Illinois start-up company iCyt. For those of you not familiar with iCyt, they are developers of the 4-headed sorting monster, the Reflection, and more recently, the Synergy (a sorter/analyzer hybrid), and now the Eclipse (a bench-top analyzer capable of doing particle sizing, absolute counts, multi-well sampling, and multi-color analysis). iCyt has certainly made a name for itself by bringing new and exciting technology to a field overpopulated with BD instruments and clones of BD instruments, and it looks like companies like Sony have taken notice. There was no way iCyt was selling enough instruments, and generating enough capital to do the development it wanted, so this acquisition is probably the best thing that could have happened to them. Gary Durack, founded and President of iCyt (and the former Director of the flow core at U of I) had this to say about the new acquisition, "As a Sony company, iCyt will be able to leverage Sony’s global resources to deliver a variety of innovative solutions to the cell analysis market. This is truly a win for iCyt, its customers, and all who will benefit from these advances." Sony desperately wants to enter the technology side of biomedical research where they feel they can make an immediate impact due to their $79 billion in annual revenue. Keiji Kimura, EVP of Sony had this to say: "iCyt’s experience and technologies will be valuable assets for Sony as it expands into this new business domain. We are confident that this acquisition will accelerate the development of Sony’s flow cytometry business by combining Sony’s expertise in the manufacturing of consumer products with the technological assets of iCyt."
I cannot wait to see the 1st flow cytometer controlled by the PSP motion controller. I mean, it had to happen sooner or later. Kids today (allow me to put on my "old man" hat for a minute) expect everything to work like a video game/social network. So why can't flow cytometers, and for that matter, all biomedical research technologies, operate like every other modern technology. Isn't scientific collaboration just a guise for social networking? Where is the Facebook of science? Why can I not run a flow cytometer with 5 of my "friends" (colleagues-represented by their avatar on screen) assisting me in picking out the important pieces of data? I know some of these things exist in some ways, but if it's not integrating seamlessly into the technology, no one will use it. This is where a company like Sony can come in and make lots of waves. They already know how to do all this stuff; think PSP marketplace, but for flow reagents, or analysis scripts, or cool software hacks. 3rd party, open-source geeks (me included) will jump all over this stuff.
One other area of importance is the whole idea of low-cost CD4 counts for areas of the developing world afflicted by HIV/AIDS. I know this is something that Gary Durack is already pursuing (see Cytometry for Life), perhaps now with a global company with lots of money and lots of influence around the world, a new integrated system that can truly allow for low-cost CD4 counts can be developed. I can't wait to see what comes out of this new marriage.
I cannot wait to see the 1st flow cytometer controlled by the PSP motion controller. I mean, it had to happen sooner or later. Kids today (allow me to put on my "old man" hat for a minute) expect everything to work like a video game/social network. So why can't flow cytometers, and for that matter, all biomedical research technologies, operate like every other modern technology. Isn't scientific collaboration just a guise for social networking? Where is the Facebook of science? Why can I not run a flow cytometer with 5 of my "friends" (colleagues-represented by their avatar on screen) assisting me in picking out the important pieces of data? I know some of these things exist in some ways, but if it's not integrating seamlessly into the technology, no one will use it. This is where a company like Sony can come in and make lots of waves. They already know how to do all this stuff; think PSP marketplace, but for flow reagents, or analysis scripts, or cool software hacks. 3rd party, open-source geeks (me included) will jump all over this stuff.
One other area of importance is the whole idea of low-cost CD4 counts for areas of the developing world afflicted by HIV/AIDS. I know this is something that Gary Durack is already pursuing (see Cytometry for Life), perhaps now with a global company with lots of money and lots of influence around the world, a new integrated system that can truly allow for low-cost CD4 counts can be developed. I can't wait to see what comes out of this new marriage.
Friday, April 16, 2010
ISX Fluorescence Intensity versus FCM

One of the first questions people ask me about the ISX is how does the fluorescence intensity compare to a standard flow cytometer (FCM). In a quick attempt to partially answer that question, I ran some BD Calibrite beads on our LSRII-Orange and our ISX, and have posted the results in the attached slide. I ran PerCP beads in hopes of demonstrating that the high laser power and long illumination times on the ISX would photobleach the signal (which it did). I also ran FITC, PE, and APC beads as sort of a general comparison of commonly used fluors. The results are fairly nice. FITC is nearly identical on the 2 instruments, PE was brighter on the FCM (probably due to filter ranges), and APC was really bright on the ISX (due to the zero background fluorescence in this channel). The other thing to test was how well the IDEAS software compensated the spillover. For FITC and PE, it was fine. However, because the PerCP was so dim in the PerCP channel, (APC was actually brighter in the PerCP channel than PerCP was) compensating those two took quite a bit of trial and error on my part. I manually adjusted the comp values until it looked acceptable. The resulting plot pretty much explains it all. You'd be hard pressed to easily identify APC, PerCP double positives in this example (luckily there were none, so no problem). Anyway, just wanted to give you a quick peak into how things may match up when trying to set up your color choices for the ISX.
Wednesday, April 14, 2010
Biolegend's Kelly Lundsten in tha house
We are hosting a seminar on multicolor cytometry next Thursday and we think it should be an absolute blast. We got fluorescent chemistry specialist Kelly Lundsten from Biolegend to come over and talk about the different factors involved in choosing the right panel of fluorochromes in a flow cytometry experiment. For example, she’ll cover the level of antigen expression, with signal:background of the fluor and detection channel, how to categorize antigens to minimize the need for multiple fluorophore versions of the same antibodies, etc. There will be an example of a 10 color assay optimization and the use of FMO controls for gate placement. She will also cover the ins and outs of the different dye chemistries too, organic dyes, proteins and nanocrystals from both a theoretical and practical perspective. Seriously, ask her about nanocrystals. Then try to interrupt her. Fun.
The seminar will be held on:
Thursday April 22nd 2h30PM to 4PM
KCBD Auditorium (room 1103)
Refreshment and snacks will be served.
This should be a very cool talk. Hope to see you there. Let me know if you have any questions.
The seminar will be held on:
Thursday April 22nd 2h30PM to 4PM
KCBD Auditorium (room 1103)
Refreshment and snacks will be served.
This should be a very cool talk. Hope to see you there. Let me know if you have any questions.
Monday, April 12, 2010
Easy FTP data Transfer
You will now see an icon on the desktop of each "windows-running" computer that says "Upload data to FTP Server." You can begin using this to transfer your data files from the acquisition computer to the Data Server. You can then log into the FTP server from your desktop computer and download the data files to your PC. Please note that the Data Server is NOT a data storage server, it is simply a means for transferring your data to your computer without having to use removable media (USB drives, CDs, etc...). You can upload your data to the server from the Macs as well, but you will not be using the Fling application. We will just map the directory on the FTP server to an alias on the desktop. Detailed instructions will be mailed to the list, if you do not receive these emails, send us an email directly or call the lab.
Now that's more like it. ISX Data Samples
We've had the opportunity to run a few real experiments on the ISX over the past week or so, and the data we're collecting seem quite interesting. We've performed the following experiments: NFkB translocation, GFP Foci Counting resulting from DNA Damage, Ploidy Analysis using CEN Reference Cells, and Cell Cycle. I've attached a few images of some compiled .ppt slides. They should be pretty self explanatory, but here's a quick run-down.
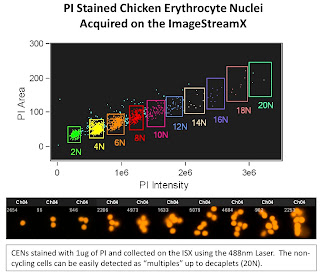
The 1st slide is looking at Chicken Erythrocyte Nuclei which is typically used as a reference peak when doing DNA ploidy analysis. These nuclei tend to pack closely together yielding discrete peaks at 2N (1 nuclei), 4N (2 nuclei), 6N (3 nuclei) and so on. The one interesting thing to note here is the ability of the ISX to display up to 20N (decaplets) on a linear scale. This gives you an idea of the dynamic range possible with this system. On a traditional Flow Cytometer, you'd only be able to display up to 8N on a linear scale due to the narrow dynamic range.
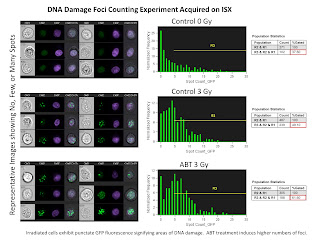
The second slide is a Nuclear Foci Counting experiment. For this we used the EDF filter (Extended Depth of Field) in order to get all foci in 3 dimensions in focus at the same time. The remarkable thing here is the ability for the IDEAS software to locate and count the number of foci inside the nucleus anywhere from 1 foci up to 25 foci.
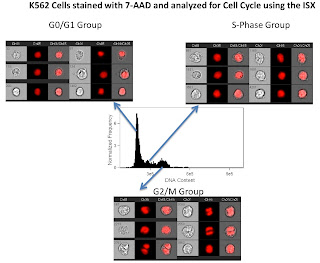
The third slide is a simple cell cycle analysis. As you can see this looks much better than my 1st attempt. What's really neat, is that using a few more morphology parameters such as the "centroid" feature, we could easily separate out the late mitotic cells (easily discernible from the images in the figure) from the G2 cells. Of course we could always add in a marker or two to label the phospho-Histones to make that process easier, but you get the idea.
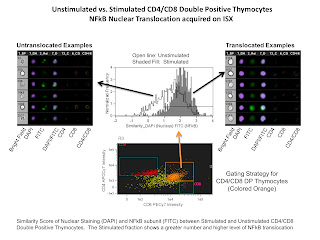
The last slide is of an NFkB tranlocation experiment. The analysis involved here compared the nuclear staining DAPI with the NFkB translocating subunit stained with FITC. If the staining is similar (i.e. translocated NFkB) a higher similarity score would be given, and if the staining is anti-similar (non-translocated NFkB) a low or negative score would be given. In this example we are simply comparing stimulated versus non-stimulated samples demonstrating a higher level of nuclear translocation of NFkB in the stimulated sample.
So, as you can see, very interesting data is being collected. If you have any questions, or want to try something, give us a call. Remember there is NO USAGE FEE on this instrument for a limited time!
The 1st slide is looking at Chicken Erythrocyte Nuclei which is typically used as a reference peak when doing DNA ploidy analysis. These nuclei tend to pack closely together yielding discrete peaks at 2N (1 nuclei), 4N (2 nuclei), 6N (3 nuclei) and so on. The one interesting thing to note here is the ability of the ISX to display up to 20N (decaplets) on a linear scale. This gives you an idea of the dynamic range possible with this system. On a traditional Flow Cytometer, you'd only be able to display up to 8N on a linear scale due to the narrow dynamic range.

The second slide is a Nuclear Foci Counting experiment. For this we used the EDF filter (Extended Depth of Field) in order to get all foci in 3 dimensions in focus at the same time. The remarkable thing here is the ability for the IDEAS software to locate and count the number of foci inside the nucleus anywhere from 1 foci up to 25 foci.

The third slide is a simple cell cycle analysis. As you can see this looks much better than my 1st attempt. What's really neat, is that using a few more morphology parameters such as the "centroid" feature, we could easily separate out the late mitotic cells (easily discernible from the images in the figure) from the G2 cells. Of course we could always add in a marker or two to label the phospho-Histones to make that process easier, but you get the idea.

The last slide is of an NFkB tranlocation experiment. The analysis involved here compared the nuclear staining DAPI with the NFkB translocating subunit stained with FITC. If the staining is similar (i.e. translocated NFkB) a higher similarity score would be given, and if the staining is anti-similar (non-translocated NFkB) a low or negative score would be given. In this example we are simply comparing stimulated versus non-stimulated samples demonstrating a higher level of nuclear translocation of NFkB in the stimulated sample.
So, as you can see, very interesting data is being collected. If you have any questions, or want to try something, give us a call. Remember there is NO USAGE FEE on this instrument for a limited time!
Friday, March 26, 2010
It ain't pretty, but it works...
I'll probably look back at this some time very soon and realize how bad this really is, but I figured I'd post it anyway. What we have here is the 1st data file collected on the ISX. I basically took the field service guys' test cells (which happened to be fixed K562s) threw a few microliters of PI on them and stuck it on the instrument. No incubation, no RNAse, no knowledge of how the cells were actually fixed (PFA, Methanol, other), no real clue on how to operate the instrument whatsoever. So, you can sort of understand my mild level of excitement that I actually got a somewhat discernible cell cycle profile. I collected the data using the Inspire software, and the shot it over to the analysis computer with the IDEAS program and played around a bit. I did a few screen grabs and made a layout in paint (that's why the resolution of the images is so poor) and displayed it below. If you squint really hard, you can see a G1, G2 and S-phase profile, and then I grabbed a representative image from those 3 groups to confirm they were actually G2 cells. I was even able to do a pretty good job of excluding aggregates. So there you go folks, it works. I guess we'll keep it.


Wednesday, March 24, 2010
ImageStream. Installed.
It seems like years ago we were racing to put together our S-10 to purchase an ImageStream, and yet, it's only been 1 year (almost to the day!). I guess the anticipation has made the wait that much more excruciating. Well, wait no more, the ImageStream is just about installed. The engineers from Amnis arrived on Monday to uncrate and assemble the surprisingly small ImageStream. I should just clarify something right off the bat. The instrument we have is the current generation ImageStream X (not sure what the X means, maybe it stands for X-treme - remember that tag line from the MoFlo XDP...a little dramatic if you ask me). Anyway, I've become fond of the abbreviation, ISX, so that's what I'll be referring to it as. Out ISX has quite the spec sheet; 4 lasers (405nm, 488nm, 561nm, 658nm), two 6-channel CCDs (up to 10 colors, Brightfield and Darkfield), the multimag (magnification from 20X - 60X), and the EDF (extended depth of field). I'm very anxious to get my hands on it. If you'd like to see it in its pre-Ryan Duggan, pristine state, you'd better stop by and take a look at it soon, because one I'm let loose, I have a hard time leaving things alone. I'll try not to break anything for at least a few weeks. We'll be getting our training next week, and then we'll probably start opening it up to users the following week (April 5th). If you think you're ready to go with something on this instrument right away, please contact me and I'll get you on the list ahead of posting the instrument on the scheduler. If you have no idea what the ISX is, check it out here.
Data Transfer post USB
I'm over USB for data transfer. There's the possibility of losing the drive altogether or it becoming corrupt, or in the case of recent happenings, it could get infected with a virus and then transmit said virus to every computer you plug it into. CDs, DVDs, BDs, may not be as hard to use, but it's still physical media that you need to label and store (away from heat and light, hopefully). So, we got to thinking, why can't we do without physical media altogether. We, in the flow lab IM or email ourselves data all the time. Sure there's a limit on the total byte size, but many of our experiments involve small numbers of tubes/small numbers of data points. Enter, the ImageStream. The data from the ImageStream (btw, it's being installed this week...more on that later) is going to be much larger, so when I was writing the grant, I put in some funds to get a full-out server that would allow us to at least FTP data to and from. If I'm already setting that up for the ImageStream, we should be able to use it for all the instruments, and manage data sans removable media across-the-board. So, over the next few weeks, we will be implementing this FTP to Server strategy on all the instruments. The LSRIIs were the test case, and they seem to be working well. I'll be outlining some detailed instructions in the next posts, but basically once you export your FCS files in the normal fashion, just zip the folder and drag it onto the upload shortcut icon the instrument desktop. Then, go to your computer and download your zipped folder of data. For the most part it should be pretty painless. The only difficult part is having an FTP client on your computer that is fairly easy to use. We will be recommending the free, Filezilla program. It runs on Mac, Windows, and Linux, so we can pretty much have 1 set of instructions for everyone, and will be able to troubleshoot, to some degree, problems you may have. For some of you who use other cores on campus (e.g. DNA Sequencing Core) this should be very familiar to you already. If you don't want to use a standalone FTP app, you can set up a handy "network place" on windows, or directly connect to the server on a mac (general instructions to follow). There's always the possibility of using a browser for this, but I'm not a fan, so won't even give instructions for doing that. I will send an email out to the list with the details of the server (server path, usernames, passwords, etc...), and then check back here to get instructions on how to proceed. If you're familiar with using FTP, then the server path, username, and password should be all you need.
Tuesday, January 5, 2010
1-on-1 FlowJo Training Slots
We will have a FlowJo specialist from Treestar out on 1/14/10 and 1/15/10 to assist any FlowJo users with problems/questions regarding flow cytometry analysis. We have set up a resource on the Flow Facility's online instrument scheduler called FlowJo Training. The resource can be found under the Training Courses calendar. A read-only version of the calendar can be found here, but to reserve your slot, you'll need to log in with your username/password. This is a great opportunity for both new users of the software who may need a little personalized help getting started as well as seasoned veterans looking to delve deeper into the vast capabilities of FlowJo. You may reserve the 30 minutes slots (1 hour max) anytime up until 1/14/10. The actual training will take place in the facility's Data Analysis room in Kovler Room 038. Please include in the summary field of the reservation form a description of what you would like help with, so that the trainer can be prepared (e.g. compensation, batch processing). Also, include which version of FlowJo you are currently using (Mac 8.8.6, Windows 7.5, etc...)
Subscribe to:
Posts (Atom)